

Abstract
Complement factor H (Cfh) is a key regulator of the complement cascade and protects C57BL/6 mice from immune complex-mediated complement-dependent glomerulonephritis. In chronic serum sickness (CSS) there are increased deposits of immune complexes in the glomeruli with inflammation and a scarring phenotype. As cucurmin is an effective anti-inflammatory agent and reduces complement activation, we hypothesized that it should alleviate renal disease in this setting. To determine the effectiveness of curcumin, an apoferritin-induced CSS model in Cfh-deficient (Cfh−/−) mice was used. Curcumin treatment (30 mg/kg) given every day in parallel with apoferritin reduced glomerulonephritis and enhanced kidney function (blood urea nitrogen, 45·4 ± 7·5 versus 35·6 ± 5·1; albuminuria, 50·1 ± 7·1 versus 15·7 ± 7·1; glomerulonephritis, 2·62 + 0·25 versus 2 + 0·3, P < 0·05). In line with reduced IgG deposits in mice with CSS given curcumin, C9 deposits were reduced indicating reduced complement activation. Mice treated with curcumin had a significant reduction in the number of splenic CD19+ B cells and the ratio of CD19 : CD3 cells (P < 0·05) with no change in the T-cell population. Myeloperoxidase assay showed reduced macrophages in the kidney. However, a significant reduction in the M2 subset of splenic macrophages by apoferritin was prevented by curcumin, suggesting a protective function. Curcumin treatment reduced mRNA expression of inflammatory proteins monocyte chemoattractant protein-1 and transforming growth factor-β and matrix proteins, fibronectin, laminin and collagen. Our results clearly illustrate that curcumin reduces glomerulosclerosis, improves kidney function and could serve as a therapeutic agent during serum sickness.
Keywords: Complement factor H, curcumin, glomerulonephritis, inflammation, serum sickness
Introduction
The complement cascade is an important arm of the innate and adaptive immune system and consists of the classical, alternative and lectin pathways.1–3 The alternative pathway is continually under a low level of activation through a process called ‘tickover’ and is kept in check by regulators located along the pathway. On activation C3b is generated which then catalyses further alternative pathway activation.4 Complement factor H (Cfh) is an important regulator of this process by preventing the formation and promoting the association of the critical C3 convertase enzyme.5 Therefore, it is not surprising, given that factor H is a major fluid phase complement regulator, that abnormalities in this protein are associated with several diseases, including atypical haemolytic uraemic syndrome and dense deposit disease.6,7 The complement factor H deficient (Cfh−/−) mice develop renal disease with features of human pathology8 and are used as the mouse model for dense deposit disease.
Factor H in rodents was also shown to participate in the trafficking of immune complexes, functioning as the immune adherence receptor similar to CR1 in humans.9 Chronic serum sickness (CSS) is a good simulation of human immune-complex-mediated glomerulonephritis. It can be induced using a number of immunization protocols10,11 and the resulting immune complexes stimulate the complement cascade leading to glomerular disease.12,13 Depletion of complement with cobra venom factor or selective inhibition of C5 reduces proteinuria, indicating the important role of complement in this setting.14–16 Stilmant et al. developed a mouse model of CSS induced by repetitive immunization with horse spleen ferritin17 with a strain and dose dependence. Our laboratory has shown that apoferritin immunization in the absence of Cfh converted the normally resistant C57BL/6 mice to be susceptible to immune-complex-mediated disease.18 We have also shown that Cfh on platelets and podocytes is needed for proper systemic and intraglomerular immune complex processing, whereas plasma Cfh is essential to prevent disease development.19
Lymphocytes such as B and T cells, dendritic cells and macrophages play a key role in the pathogenesis of immune-mediated glomerulonephritis.20,21 B cells, CD4+ cells and macrophages could mediate the delayed-type hypersensitivity mechanism of crescent formation in experimental glomerulonephritis.20 B cells are converted to plasma cells, which generate antibodies.22 B cells express cell surface molecules including CD19, which regulate antigen responses, development, tolerance induction, proliferation and differentiation. The antibodies deposited can lead to inflammation, which can attract macrophages and trigger their polarization into pro- (M1) or anti-inflammatory (M2) subtypes. The heterogeneity of the M2 macrophages is reflected in their subdivision into M2a, M2b and M2c cells.23 The subsets, M2a, b and c range from those that confer immunity to those that participate in tissue repair. Macrophages contain myeloperoxidase (MPO), an enzyme that is implicated in the pathogenesis of kidney disease because both MPO itself and its products have been demonstrated in diseased renal tissue.24 Macrophages are also the predominant cell type responsible for the generation of transforming growth factor-β (TGF-β) and thereby the development of fibrosis and progressive fibrotic scarring.25,26
The Asian spice, curcumin (diferuloylmethane, CMN) is an active principle component isolated from the rhizome of Curcuma longa27 and displays anti-inflammatory and anti-oxidant properties.28 Curcumin was shown to regulate both the classical and alternative complement pathways.29,30 No previous study has examined the use of CMN in the treatment of complement-mediated glomerular diseases, so this study investigated the effect of CMN on kidney injury during serum sickness in Cfh-deficient mice.
To understand the effect of CMN in dense deposit disease and immune complex-mediated diseases, we administered CMN to Cfh−/− mice with CSS in which immune complex processing and complement activation are key. For the first time we show that CMN can alleviate disease symptoms in Cfh−/− mice and can potentially be used as an adjuvant therapy in this setting.
Materials and methods
Reagents
Curcumin was purchased from Calbiochem (Billerica, MA). Horse spleen apoferritin was procured from (Calzyme Laboratories, San Luis Obispo, CA).31 Sybr green and other PCR reagents were from Applied Biosystems Inc. (Foster City, CA). Oligonucleotides were generated by Integrated DNA Technologies (Coralville, IA). C3 and IgG antibodies were obtained from (Cappel, MP Biomedicals, Solon, OH). C9 antibody was a kind gift from Dr Scott Barnum (Birmingham, AL). All other antibodies used for FACS analysis were bought from BD-Pharmingen (Franklin Lakes, NJ) unless indicated otherwise. All reagents unless stated differently were from Sigma Chemical Co. (St Louis, MO).
Experimental protocol in Cfh−/− mice
The Cfh−/−8 mice on the C57BL/6 strain used were generously provided by Dr Marina Botto, Imperial College, London, UK. Cfh−/− mice were divided into three groups of six mice each. Group 1: mice were treated with saline and served as controls; Group 2: immune complex glomerulonephritis was induced by immunizing 6- to 8-week-old male Cfh−/− mice (n = 6) for 5 weeks with a daily intraperitoneal dose of 4 mg horse spleen apoferritin, as described previously.32 Group 3: mice were treated intraperitoneally with apoferritin and CMN obtained from Calbiochem,33 30 mg/kg. As CMN is non-polar it was suspended in 25 μl DMSO. Control Cfh−/− mice (n = 5) were treated identically, except they received 25 μl vehicle alone. These animals were killed and a comprehensive assessment of disease phenotype was performed, as we have described previously in this model.31 All animal procedures were approved by the University of Chicago Institutional Animal Care and Use Committee.
Kidney function
Blood urea nitrogen (BUN) concentrations were detected with a Beckman Autoanalyzer (Beckman Coulter, Fullerton, CA). Serum creatinine was measured with Stanbio Creatinine Procedure No. 0400, (Stanbio Laboratory, Boerne, TX). Urinary albumin concentrations were measured with a mouse albumin ELISA kit (Bethyl Laboratories, Montgomery, TX) and normalized to creatinine concentrations in the same urine.
Serum C3 levels, and circulating immune complex levels were determined by ELISA as described previously.34 For serum C3 ELISA, plates were coated with goat anti-mouse C3 (Cappel Laboratories, Durham, NC). Serially diluted serum samples were added followed by horseradish peroxidase-goat anti-mouse C3 (Cappel Laboratories). Results are expressed as relative optical density at 450 nm values adjusted for standard wild-type C57BL/6 mouse serum samples included in all. The anti-mouse C3 antibody identifies native intact C3 along with the cleavage fragments iC3b and its component C3c to some extent.
To measure circulating immune complex levels, 96-well plates were coated with human C1q (Quidel, San Diego, CA). Serum samples were serially diluted and incubated for 2 hr at room temperature, followed by horseradish peroxidase-goat anti-mouse IgG (Jackson ImmunoResearch Laboratories, West Grove, PA) and o-Phenylenediamine dihydrochloride (OPD) peroxidase substrate (Sigma). Sera from several apoferritin treated Cfh−/− mice were pooled and served as controls. Circulating immune complex levels were quantified by plotting against the standard curve.
FACS analysis
To enumerate and characterize the peripheral leucocytes and splenic cells, flow cytometry was performed. Splenocytes were isolated according to standard techniques and treated with ACK lysis solution (0·15 m NH4Cl, 1 mm KHCO3 and 0·1 mm Na2EDTA, pH 7·2) for 5 min to deplete red blood cells. Cells were washed and resuspended in FACS buffer (2% fetal bovine serum, 0·05% NaN3 and 2 mm EDTA in PBS) to a final concentration of 107/ml. Blood was collected in EDTA followed by red blood cell lysis and cells were resuspended in FACS buffer. Then, 100 μl (106 cells) of the splenocytes or peripheral blood cells were incubated with Fc block (1 μg monoclonal antibody 2.4G2) for 5 min. Cells were simultaneously stained for 1 hr with the following monoclonal antibodies: Brilliant Violet 421 αCD3 (145C211), allophycocyanin-Cy7 αCD19 (6D5), Alexafluor 647 αCD8 (KT15) (Molecular Probes, Inc., Eugene, OR), FITC αCD4 (W3/25), V500 αCD11b (M1/70), Peridinin chlorophyll protein-Cy5.5 αLy6C (HK1.4), phycoerythrin (PE) αCCR2 (475301), PE-Cy5 αCD62L (MEL-14), PE-Cy7 αF4/80 (BM8) (Biolegend, San Diego, CA), Alexafluor 700 αCD11c (N418). All antibodies were procured from BD PharMingen (San Diego, CA). Samples were washed and resuspended in FACS buffer and ran on an LSRII instrument (BD Biosciences, Franklin Lakes, NJ). Analysis was performed with FlowJo software (Tree Star, Inc., Ashland, OR).
Histology
To evaluate renal pathological changes, kidney tissues were fixed in 4% paraformaldehyde and embedded in paraffin. Four-micrometer sections were stained with periodic acid-Schiff. Slides were scored in a blinded manner by a renal pathologist (AC) for the extent of glomerulonephritis and interstitial nephritis using scales of 0–4 (in increments of 0·5) as described previously.31
Kidney homogenate
The kidney was cut and immediately frozen in liquid nitrogen and kept at − 70° until use. The frozen kidney was ground to a powder and then mixed in ice-cold HEPES buffer (10 mm HEPES, 0·2% Triton X-100, 50 mm NaCl, 0·5 mm sucrose, 0·1 mm EDTA, protease and phosphatase inhibitors) and homogenized with an ice-chilled dounce homogenizer at 4°. The homogenate was stored as aliquots at − 70°.
Macrophages
Macrophages infiltrating the kidney were assessed using the myeloperoxidase detection kit from Cell Technology Inc. (Mountain View, CA), according to the manufacturer's instructions. In brief, homogenates were prepared and centrifuged at 12 000 g at 4°. The pellet was solubilized and subjected to freeze–thaw cycles. The reaction cocktail was prepared according to instructions and added to the samples. N-Ethylmaleimide (Sigma Cat.# E1271) was added to prevent interference due to the presence of glutathione and other reductants. The homogenates were incubated with 20 mm 3-amino-1,2,4-triazole (Sigma Cat.# A8056) for 60 min before running the assay to inhibit catalase activity, if present. Fluorescence was measured at excitation 530–570 nm and emission at 590–600 nm.
Immunofluorescence microscopy
For immunofluorescence (IF) microscopy, 4-μm cryostat sections were fixed in 1 : 1 ether : ethanol and stained with FITC-anti-mouse C3 (Cappel Pharmaceuticals, Aurora, OH), Alexa 647-anti mouse IgG and rabbit anti-mouse C9 followed by Alexa 594-anti-rabbit antibody (Molecular Probes). Primary antibodies were used at a dilution of 1 : 100 and secondary antibodies at 1 : 300. Slides were viewed with an Olympus BX-60 IF microscope (Carter Valley, PA). Representative photomicrographs were taken at identical settings with a Hamamatsu EM-CCD camera (Bridgewater, NJ). Mesangial staining intensity scores from 0 to 4 were assigned to the individual samples by observers who were masked to their origin. All assays included negative controls where the primary antibody was omitted.
Quantitative real-time RT-PCR analysis
RNA was isolated from kidneys using TRIzol reagent (Life Technologies, Grand Island, NY) as described previously.35 Quantitative RT-PCR was performed as described previously. Real-time quantitative RT-PCR was performed for collagen, fibronectin, laminin, keratinocyte-derived cytokine (KC), CXCL2/macrophage inflammatory protein 2 (MIP-2), TGF-β, intercellular adhesion molecule 1 (ICAM-1), monocyte chemoattractant protein 1 (MCP-1) and C3. All traces of genomic DNA were eliminated with RQ1 DNase (Promega, Madison, WI) at 37° for 30 min. The cDNA was produced from renal cortical RNA using a commercial kit (Superscript III; Invitrogen). Quantitative RT-PCR was performed with the QuantiTect SYBR Green RT-PCR Kit (Qiagen, Valencia, CA) using an Applied Biosystems 7300 Real Time PCR System and the SybrGreen intercalating dye method with Hot-Star DNA polymerase (PE Applied Biosystems) according to the manufacturer's instructions. PCR was conducted with a hot start at 95° (5 min), followed by 45 cycles of 95° for 15 seconds and 60° for 30 seconds. For each sample, the number of cycles required to generate a given threshold signal (Ct) was recorded. Expression data were normalized to 18S RNA measured contemporaneously from the same samples. Primers were synthesized by Integrated DNA Technologies. The sequences of primers/probes are given below.
Primers used in quantitative RT-PCR
| Genes | Primer 1 (Forward) | Primer 2 (Reverse) |
|---|---|---|
| 18S | 5′-atggccgttcttagttggtg-3′ | 5′-cgctgagccagtcagtgtag-3′ |
| MCP-1 | 5′-accacagtccatgccatcac-3′ | 5′-ttgaggtggttgtggaaaag-3′ |
| MIP-2 | 5′-caccaaccaccaggctac-3′ | 5′-gcccttgagagtggctatga-3′ |
| ICAM-1 | 5′-cgcaagtccaattcacactga-3′ | 5′-cagagcggcagagcaaaag-3′ |
| KC | 5′-catggctgggattcacctc-3′ | 5′-tctgtcttctttctccgttacttg-3′ |
| TGF-β1 | 5′-ttgcttcagctccacagaga-3′ | 5′-tggttgtagagggcaaggac-3′ |
| C3 | 5′-atcagccacatcaagtgcag-3′ | 5′-ctgtgaatgccccaagttct-3′ |
Statistical analysis
Numerical data from all experiments were first analysed using the ‘graphical summary’ function in Minitab 15 (State College, PA) to determine data normality (Anderson–Darling test) and 95% confidence intervals for mean, median and SD. Parametric and non-parametric data were analysed by one-way analysis of variance and Kruskal–Wallis tests, respectively. Comparisons within each group for a given parameter were made using paired Student's t-tests; P < 0·05 was considered statistically significant.
Results
Curcumin protects against renal failure in Cfh-deficient mice with CSS
Studies were performed to examine the effect of curcumin on the functional and histopathological features of glomerulonephritis in Cf−/− mice. Mice with 5 weeks of apoferritin treatment developed albuminuria (48·7 ± 6·4 versus 13·8 ± 1·5; P < 0·05) as seen in our studies earlier. Comparable to our earlier studies, BUN levels were elevated compared with control (45·4 ± 7·5 versus; 26·7 ± 2·6; P < 0·05). Curcumin treatment reduced both albuminuria (15·7 ± 7·1; P < 0·05) and the level of BUN (30·2 ± 2·9 mg/dl, P < 0·05 compared with apoferritin treated; Table 1) in Cfh−/− mice, indicating that it was protective of kidney function in this setting. This was further substantiated by the significant reduction in serum creatinine in mice treated with CMN compared with those treated with apoferritin alone (Fig. 1c).
Table 1.
Effect of curcumin on renal function in immune complex glomerulonephritis
| Immunogen | BUN (mg/dl) | Albuminuria (g/mg creatinine) | n |
|---|---|---|---|
| Saline | 26·7 ± 2·6 | 13·8 ± 1·5 | 6 |
| Apoferritin | 45·4 ± 7·5** | 48·7 ± 6·4** | 6 |
| Apoferritin + CMN | 30·2 ± 2·9* | 15·7 ± 7·1* | 6 |
BUN (blood urea nitrogen) and albuminuria levels were measured after 5 weeks of daily immunization with either DMSO, apoferritin or apoferritin + curcumin (CMN) in DMSO. Data are mean ± SD
P < 0·05;
P < 0·01.
Figure 1.

Curcumin (CMN) alleviates glomerulonephritis in Complement factor H-deficient (Cfh−/−) mice with chronic serum sickness. (a) Representative periodic acid-Schiff-stained kidneys from C57BL/6 Cfh−/− mice treated with saline, apoferritin or CMN + apoferritin. Hypercellularity and sclerosis are evident in apoferritin-treated mice. The pathology is significantly reduced when the mice were treated with CMN. (b) Blinded scoring for the extent of glomerulonephritis is shown. (c) Serum creatinine was measured in the different groups as described in the Materials and methods. Creatinine levels were significantly increased in apoferritin-treated mice compared with controls. This increase was prevented by CMN treatment. **, P < 0·01; *P < 0·05.
Curcumin alleviates glomerulonephritis in CfH-deficient mice with CSS
Formalin-fixed kidneys were embedded in paraffin wax and sections were stained with periodic acid-Schiff (Fig. 1a). Control Cfh−/− mice receiving saline had mild to no evident glomerular pathology, while Cfh−/− mice immunized with apoferritin developed significant glomerulonephritis, characterized by diffuse hypercellularity of the glomerular tufts as observed earlier. Sections were read by the pathologist in a blinded fashion to assess glomerular pathology (Fig. 1b). Treatment with CMN protected Cfh−/− mice with CSS from renal pathology as the glomeruli looked closer to normal. Figure 1 shows these features in three representative examples of histological changes in each group.
We were then interested in whether CMN affected glomerular localization of C3 and IgG-containing immune complexes in this experimental serum sickness model (Fig. 2). As seen in all our earlier studies, immunofluorescence microscopy showed substantial linear C3 staining along glomerular capillary walls in Cfh−/− C57BL/6 mice by 8 weeks of age, which continued to maintain the same profile after 5 weeks of apoferritin and even after CMN treatment (Fig. 2). Although the Cfh−/− mice have marked consumption of circulating C3, these animals continued to deposit C3 within the kidney. This C3 may be derived from C3 produced intrinsically by glomerular epithelial and mesangial cells.18,36 In line with our earlier observations, there was minimal IgG in saline-treated mice and substantial IgG deposits in mice with CSS.18 In Cfh−/− mice with serum sickness, there was a qualitative and quantitative difference of IgG deposits (as scored in Fig. 2b), in that Cfh−/− animals had more intense mesangial staining as well as greater involvement in peripheral capillary loops. The pattern of IgG and C3 deposition was different, so there was limited overlap and in regions of extensive deposits it even seemed to displace C3 (Fig. 2, arrow). Treatment with CMN reduced IgG deposits in these mice. To determine whether the late complement pathway (C5b-9) was affected in this setting, we determined C9 deposition by immunofluorescence. In Cfh−/− mice with serum sickness, there was a significant increase in C9 deposits compared with saline-treated mice. Similar to the C3, IgG seemed to displace the C9 to the periphery. In CMN-treated Cfh−/− mice with serum sickness, C9 deposits were reduced and, in line with the reduced IgG, were found distributed evenly across the glomerulus.
Figure 2.

Curcumin (CMN) reduces IgG deposits in Complement factor H-deficient (CfH−/−) mice with chronic serum sickness. Merged representative immunofluorescence photomicrographs are shown for IgG (blue), C3 (green) and C9 (red). C3 had the characteristic linear deposits along the capillary walls presenting a wire loop appearance, whereas IgG deposits were mesangial. Very little C9 was present in the controls and was significantly increased in mice treated with apoferritin. C3 remained comparable in mice treated with CMN whereas IgG and C9 deposits were reduced by treatment. (b) Blinded scoring for the extent of IgG deposition is shown. **, P < 0·01; *P < 0·05.
Curcumin alters immunological features in Cfh−/− mice with CSS
Humoral immunity was assessed in the three groups of Cfh−/− mice, those treated with saline, apoferritin and CMN + apoferritin. As demonstrated in our earlier studies, Cfh deficiency leads to increased C3 consumption and therefore to reduced C3 in circulation. This feature was further aggravated by CMN treatment with C3 non-detectable in plasma (results not shown). To understand the effect of factor H and CMN on the synthesis of C3, mRNA expression was assessed. Interestingly, C3 synthesis is reduced significantly both in the liver (55·6 ± 9·8% reduction, P = 0·003) and the kidney (60·8 ± 1·8% reduction, P = 0·016) in mice treated with apoferritin compared with control (Fig. 3). However, the synthesis of kidney C3 mRNA occurs 30 times slower than synthesis of liver C3 mRNA. Curcumin reduces the decrease and improves C3 synthesis in this setting. Normal C57BL/6 mice were used as controls. On the other hand, circulating immune complexes were increased by apoferritin treatment. In addition to apoferritin, when the mice were given CMN the circulating immune complexes were significantly reduced (Fig. 4), P < 0·05.
Figure 3.

Curcumin (CMN) protects C3 mRNA expression in liver and kidney. C3 mRNA expression was assessed in the liver and kidney of mice treated with apoferritin, apoferritin and CMN, or saline by quantitative real-time PCR as described in Materials and methods. C3 synthesis was significantly decreased in both liver and kidney when treated with apoferritin. Curcumin prevented this change in both organs. Values are expressed as mean ± SD *P < 0·05.
Figure 4.

Curcumin (CMN) reduces circulating immune complexes in Complement factor H-deficient (Cfh−/−) mice with chronic serum sickness. Apoferritin treatment increased circulating immune complexes in these mice which is also prevented by CMN treatment. On administering CMN, the immune complexes in circulation were significantly reduced. Each value represents individual animals. *P < 0·05.
Effect of curcumin on cellular autoimmunity was evaluated in Cfh-deficient mice with CSS
Lymphocytes isolated from spleens and blood were analysed by flow cytometry. The percentage of blood CD3+, CD11b+ cells remained static (results not shown). FACS analysis on splenic cells also demonstrated that mice treated with apoferritin or apoferritin + CMN do not differ in their CD4+ and CD8+ populations (Fig. 5a). However, % of CD19+ cells (Fig. 5b), activated B cells (CD19+ CD62L) and the ratio of CD19 : CD3 were significantly reduced by CMN treatment (Table 2). The monocyte population was not altered by the treatments, except the alternate, deactivated monocytes (CD11b+ Gr1-CCR2+), which were significantly reduced by apoferritin treatment (Fig. 5c). Treatment with CMN prevented the decrease in this subset of monocytes, suggesting a protective function in immune complex glomerulonephritis.
Figure 5.

Influence of curcumin (CMN) on the relative percentage of splenocytes in different subsets. Percentage of the individual cells within the total CD11b negative population is shown. (a) The percentage of CD4+ and CD8+ cells remained static compared with controls. (b) A significant decrease was observed in the % of CD19+ (B cells) when mice were treated with CMN. (c) The percentage of the individual cells within the total CD11b+ population is shown. The percentage of M2c (Gr1-CCR2+) cells was significantly reduced compared with controls in mice with chronic serum sickness. CMN treatment prevented the reduction in this subset of macrophages. All other populations remained static compared with controls. Values are means ± SE. n = 6. *Significantly different compared with the respective control, P < 0·05.
Table 2.
Curcumin (CMN) affects splenic B cells in Complement factor H-deficient mice with chronic serum sickness
| Phenotype | Saline | Apoferritin | Apoferritin + CMN |
|---|---|---|---|
| CD3+ | 25·33 + 3·06 | 29·00 + 1·86 | 31·12 + 2·78 |
| CD4+ | 54·80 + 1·47 | 57·68 + 2·03 | 54·74 + 1·80 |
| CD8+ | 39·80 + 1·15 | 36·83 + 2·21 | 40·14 + 1·87 |
| CD4+ CD62L+ | 62·10 + 8·49 | 58·00 + 6·20 | 63·52 + 5·43 |
| CD8+ CD62L+ | 89·77 + 2·92 | 85·03 + 4·78 | 85·34 + 4·84 |
| CD19+ | 64·30 + 2·35 | 60·68 + 2·25 | 54·56 + 3·88* |
| CD19+ CD62L+ | 50·90 + 1·54 | 27·00 + 4·55* | 22·00 + 6·59* |
| CD19 : CD3 | 2·57 + 0·37 | 2·10 + 0·19 | 1·76 + 0·21* |
Shown is the percentage of the individual cells within the total CD11b negative population. Populations of splenic CD3+ cells (CD4+ and CD8+ cells) and their activated counterparts (CD4+ CD62L+, CD8+ CD62L+) remained unchanged in all three groups. CD19+ B cells were significantly reduced by CMN treatment. Data are means ± SEM.
P < 0·05.
The possibility that macrophages are important inflammatory mediators in this setting was assessed using the MPO assay.36 The MPO analysis of kidney homogenates revealed significant increase in macrophage infiltration (P > 0·05) after apoferritin treatment (Fig. 6). Macrophages infiltrating the kidney were increased by 356% in Cfh−/− mice with serum sickness compared with control, which was reduced to 156% in CMN-treated mice compared with control.
Figure 6.
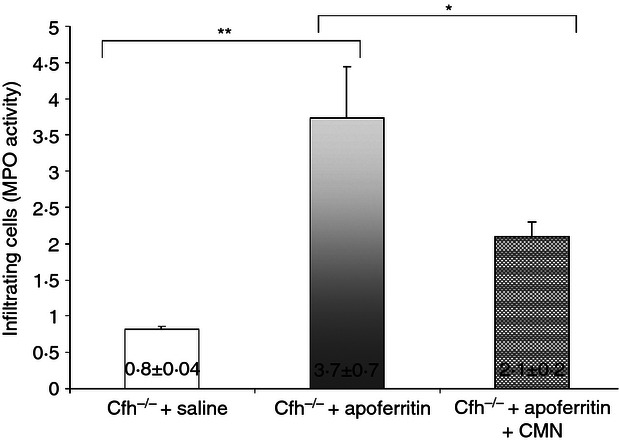
Curcumin (CMN) reduces myeloperoxidase (MPO) activity in Complement factor H-deficient (Cfh−/−) mice with chronic serum sickness. MPO activity was assessed in the kidneys of mice from the different groups as described in the Materials and methods. The fluorescent analogue that was generated from the detection reagent was measured. Values are presented as means ± SD. n = 6; **P < 0·01; *P < 0·05.
Curcumin reduces mediators of renal inflammation in Cfh−/− mice with CSS
Infiltration of inflammatory cells is regulated in part by cytokines or chemokines that modulate the activity of adhesion receptors and direct migration of cells to the tissue site. We therefore examined the effect of CMN on renal chemokine expression. Analysis of the expression of inflammatory mediators by quantitative RT-PCR indicated that mRNA expression of MCP-1, ICAM-1, TGF-β and MIP-2 were all increased in CSS whereas KC was unaltered (Table 3). Treatment with CMN prevented the increased expression of MCP-1, TGF-β and MIP-2, while ICAM-1 showed a downward trend.
Table 3.
Curcumin (CMN) reduces mRNA expression of inflammatory proteins in Complement factor H-deficient mice with chronic serum sickness
| Immunogen | MCP-1 | ICAM | TGF-β | KC | MIP-2 |
|---|---|---|---|---|---|
| Saline | 0·52 + 0·02 | 0·75 ± 0·04 | 0·63 ± 0·06 | 2·88 ± 0·42 | 1·56 ± 0·12 |
| Apoferritin | 2·02 + 0·14* | 1·92 ± 0·21* | 2·23 ± 0·76* | 3·26 ± 1·38 | 3·06 ± 1·25* |
| Apoferritin + CMN | 1·33 + 0·06* | 1·68 ± 0·43 | 0·98 ± 0·15* | 2·75 ± 0·29 | 1·75 ± 0·53* |
Expression of mRNA for the listed inflammatory proteins in renal cortices from wild-type and Complement factor H-deficient mice after 5 weeks of daily immunization with either saline, apoferritin or apoferritin + CMN was determined by quantitative RT-PCR and normalized to expression of 18S RNA from the same sample. Data are presented as Mean ± SD.
P ≤ 0·05 compared to the other two groups.MCP-1, monocyte chemoattractant protein 1; ICAM, intercellular adhesion molecule; TGF-β, transforming growth factor-β; KC, keratinocyte-derived cytokine; MIP-2, macrophage inflammatory protein 2.
Curcumin treatment prevents glomerulosclerosis in Cfh−/− mice with CSS
Consistent with our earlier studies in apoferritin-immunized Cfh−/− mice, there was an increase in mRNA for collagen IV (0·88 ± 0·04 versus 5·47 ± 0·82), fibronectin (3·08 ± 0·42 versus 6·38 ± 0·54) and laminin (0·95 ± 0·16 versus 3·00 ± 1·22) compared with controls as measured by quantitative RT-PCR (Table 4). Treatment with CMN prevented the increase in mRNA of extracellular matrix (ECM) proteins, maintaining them closer to normal.
Table 4.
Curcumin prevents increase in extracellular matrix mRNA expression in Complement factor H-deficient mice with chronic serum sickness
| Immunogen | Laminin | Fibronectin | Collagen IV |
|---|---|---|---|
| Saline | 0·95 ± 0·16 | 3·08 ± 0·42 | 0·88 ± 0·04 |
| Apoferritin | 3·00 ± 1·22* | 6·38 ± 0·54* | 5·47 ± 0·82* |
| Apoferritin + CMN | 2·56 ± 0·85 | 2·05 ± 0·29* | 1·55 ± 0·13* |
Expression of mRNA for the listed matrix components in renal cortices from wild-type and Complement factor H-deficient mice immunized with either saline, apoferritin or apoferritin + curcumin (CMN) was determined by quantitative RT–PCR and normalized to expression of 18S RNA from the same sample. Data are presented as mean ± SD.
P ≤ 0·05 compared with the other two groups.
Discussion
Curcumin, an Asian spice from the rhizome of Curcuma longa, is an inhibitor of the complement cascade and alleviates a number of immune-mediated diseases.37–39 In this study, we analysed the effect of CMN on glomerulonephritis in Cfh−/− with CSS. As functional parameters of glomerular integrity, BUN, serum creatinine and albuminuria were reduced by CMN treatment. The CMN reduced IgG deposits in the kidney in the CSS setting, which could be a result of the reduced number of CD19+ cells.40 Curcumin protects kidney function by reducing infiltrating cells, inflammation, fibronectin production and thereby fibrosis. The antifibrotic effect was evident by the reduced expression of ECM proteins.
Serum sickness is a hypersensitivity reaction where the immune complexes cause complement activation thereby leading to inflammation. Increased IgG deposits in CSS could lead to the increased infiltration of inflammatory cells. To begin to elucidate the possible mechanisms underlying inflammation in CSS, we studied the cellular autoimmunity in these mice. A decrease in splenic B cells by CMN treatment could be the reason for reduced IgG in deposits and in circulation in this setting.
The complement cascade culminates with the formation of the C5b–9 complex. In normal, untreated mice there were little to no C9 deposits, indicating that there was no complement activation. On the other hand, there was a significant increase of C9 deposits in the mice treated with apoferritin, indicating increased complement activation. In addition, the presence of C9 deposits demonstrates that the downstream complement cascade was not affected in this setting. When the mice with CSS were treated with CMN there was a reduction in C9 deposits similar to the reduction in IgG deposits.
Macrophage phenotype and function are key determinants for fibrotic scarring and resolution of injury. An interesting feature in our study is the observed decrease in the macrophage subset, M2c in CSS. The M2c cells participate in immune suppression and tissue repair and matrix remodelling.23,41 Treatment with CMN prevented the decrease in M2c cells, enabling better repair of injured tissue. Macrophages are attracted to the site of injury by MCP-1 and its receptor CCR2. Blocking this pathway was shown to reduce kidney fibrosis as this leads to reduced recruitment of M1 inflammatory macrophages.42–46 In line with this understanding, in our studies MCP-1 was increased on apoferritin treatment but was reduced by CMN treatment, which could reduce the number of activated macrophages infiltrating the kidney. In addition, reduction in infiltrating macrophages was reflected in MPO activity, a marker of macrophage activation.47 Macrophages may be the effector cells that mediate renal dysfunction because they could potentially plug the capillaries and release damaging free radicals and proteases. Levels of MPO were increased in apoferritin-treated mice, which was prevented by CMN treatment. On the one hand macrophages may have been attracted into the glomerulus by complement (e.g. C5a generated by complement activation in the absence of the regulator, complement factor H) and adhesion molecules.48,49 On the other hand, complement could regulate ICAM.50–52 The C5a receptor, CD88 is present on inflammatory cells, neutrophils and macrophages along with some tubular and glomerular cells53,54 and initiates the development of fibrosis. Macrophages stimulate mesangial cells to produce ECM proteins through TGF-β,55,56 in autocrine and paracrine fashions.57 Depletion of macrophages by irradiation decreased the gene expression of TGF-β and type IV collagen in the glomeruli of diabetic rats, suggesting a pathological role for macrophages in the increased expression of ECM proteins.58
Glomerulosclerosis is due to an excessive accumulation of ECM such as collagen IV, fibronectin and laminin, caused by increased production and/or insufficient degradation.54 Several studies suggest that complement activation may be linked with renal fibrogenesis.18,59 Our study shows that CMN is antifibrotic, associated with reduced expression of the ECM proteins, collagen, laminin and fibronectin and could be potentially acting through the complement cascade. To understand the mechanism by which these ECM proteins were turned on, we assessed the expression of TGF-β, which is widely regarded as the key fibrosis-promoting molecule. The TGF-β stimulates60–62 ECM production and is associated with several glomerular diseases.63 Expression was significantly reduced in kidneys with CSS suggesting that CMN reduced ECM protein synthesis in this setting through TGF-β. Our study shows that CMN has anti-fibrotic properties and that this effect is dependent on regulation of the complement cascade and subsequently the infiltrating cells and ECM proteins.
In brief, CMN reduced kidney disease in immune complex glomerulonephritis by preventing the reduction of alternative macrophages thereby enabling resolution and reducing number of macrophages in the kidney, expression of pro-inflammatory mRNAs (KC and MIP-2) as well as TGF-β and the ECM proteins collagen IV, laminin and fibronectin. Therefore, our data suggest that CMN has a number of beneficial effects and may be a potential adjuvant therapy in chronic human kidney disease.
Acknowledgments
We thank Dr Marina Botto (Imperial College, UK) for providing CfH-deficient mice. We express our gratitude to Bradley Hack and Rebecca Alexander for technical assistance. This work was supported by a grant from the Kidneeds Foundation (to J.J.A.), T32GM007019 (ME) and NIH R01 Grants DK041873 and DK055357 (to R.J.Q.).
Disclosures
None.
References
- 1.Campbell RD, Law SKA, Reid KBM, Sim RB. Structure, organization, and regulation of the complement genes. Annu Rev Immunol. 1988;6:161–95. doi: 10.1146/annurev.iy.06.040188.001113. [DOI] [PubMed] [Google Scholar]
- 2.Carroll MC. The role of complement and complement receptors in induction and regulation of immunity. Annu Rev Immunol. 1998;16:545–68. doi: 10.1146/annurev.immunol.16.1.545. [DOI] [PubMed] [Google Scholar]
- 3.Carroll MC. A protective role for innate immunity in autoimmune disease. Clin Immunol. 2000;95:S30–8. doi: 10.1006/clim.1999.4813. [DOI] [PubMed] [Google Scholar]
- 4.Pangburn MK, Müller-Eberhard HJ. The alternative pathway of complement. Springer Semin Immunopathol. 1984;7:163–92. doi: 10.1007/BF01893019. [DOI] [PubMed] [Google Scholar]
- 5.Alexander JJ, Quigg RJ. The simple design of complement factor H: looks can be deceiving. Mol Immunol. 2007;44:123–32. doi: 10.1016/j.molimm.2006.07.287. [DOI] [PubMed] [Google Scholar]
- 6.Smith RJ, Harris CL, Pickering MC. Dense deposit disease. Mol Immunol. 2011;48:1604–10. doi: 10.1016/j.molimm.2011.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Atkinson JP, Goodship TH. Complement factor H and the hemolytic uremic syndrome. J Exp Med. 2007;204:1245–8. doi: 10.1084/jem.20070664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pickering MC, Cook HT, Warren J, Bygrave AE, Moss J, Walport MJ, Botto M. Uncontrolled C3 activation causes membranoproliferative glomerulonephritis in mice deficient in complement factor H. Nat Genet. 2002;31:424–8. doi: 10.1038/ng912. [DOI] [PubMed] [Google Scholar]
- 9.Alexander JJ, Hack BK, Cunningham PN, Quigg RJ. A protein with characteristics of factor H is present on rodent platelets and functions as the immune adherence receptor. J Biol Chem. 2001;276:32129–35. doi: 10.1074/jbc.M101299200. [DOI] [PubMed] [Google Scholar]
- 10.Iskandar SS, Jennette JC, Wilkman AS, Becker RL. Interstrain variations in nephritogenicity of heterologous protein in mice. Lab Invest. 1982;46:344–51. [PubMed] [Google Scholar]
- 11.Iskandar SS, Gifford DR, Emancipator SN. Immune complex acute necrotizing glomerulonephritis with progression to diffuse glomerulosclerosis. A murine model. Lab Invest. 1988;59:772–9. [PubMed] [Google Scholar]
- 12.Couser WG. Mediation of immune glomerular injury. J Am Soc Nephrol. 1990;1:13–29. doi: 10.1681/ASN.V1113. [DOI] [PubMed] [Google Scholar]
- 13.Quigg RJ. Complement and autoimmune glomerular diseases. Curr Dir Autoimmun. 2004;7:165–80. doi: 10.1159/000075692. [DOI] [PubMed] [Google Scholar]
- 14.Bartolotti SR, Peters DK. Delayed removal of renal bound antigen in decomplemented rabbits with acute serum sickness. Clin Exp Immunol. 1978;32:199–206. [PMC free article] [PubMed] [Google Scholar]
- 15.Bartolotti SR, Peters DK. Complement fixation in acute serum sickness: assembly of glomerular-bound C3-convertase. Clin Exp Immunol. 1979;37:391–8. [PMC free article] [PubMed] [Google Scholar]
- 16.Furness PN, Turner DR. Chronic serum sickness glomerulonephritis: removal of glomerular antigen and electron-dense deposits is largely dependent on plasma complement. Clin Exp Immunol. 1988;74:126–30. [PMC free article] [PubMed] [Google Scholar]
- 17.Stilmant MM, Couser WG, Cotran RS. Experimental glomerulonephritis in the mouse associated with mesangial deposition of autologous ferritin immune complexes. Lab Invest. 1975;32:746–56. [PubMed] [Google Scholar]
- 18.Alexander JJ, Pickering MC, Haas M, Osawe I, Quigg RJ. Complement factor h limits immune complex deposition and prevents inflammation and scarring in glomeruli of mice with chronic serum sickness. J Am Soc Nephrol. 2005;16:52–7. doi: 10.1681/ASN.2004090778. [DOI] [PubMed] [Google Scholar]
- 19.Alexander JJ, Wang Y, Chang A, Jacob A, Minto AW, Karmegam M, Haas M, Quigg RJ. Mouse podocyte complement factor H: the functional analog to human complement receptor 1. J Am Soc Nephrol. 2007;18:1157–66. doi: 10.1681/ASN.2006101125. [DOI] [PubMed] [Google Scholar]
- 20.Ricardo SD, van Goor H, Eddy AA. Macrophage diversity in renal injury and repair. J Clin Invest. 2008;118:3522–30. doi: 10.1172/JCI36150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Clatworthy MR, Smith KG. B cells in glomerulonephritis: focus on lupus nephritis. Semin Immunopathol. 2007;29:337–53. doi: 10.1007/s00281-007-0092-1. [DOI] [PubMed] [Google Scholar]
- 22.Pillai S, Mattoo H, Cariappa A. B cells and autoimmunity. Curr Opin Immunol. 2011;23:721–31. doi: 10.1016/j.coi.2011.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mantovani A, Sica A, Locati M. Macrophage polarization comes of age. Immunity. 2005;23:344–6. doi: 10.1016/j.immuni.2005.10.001. [DOI] [PubMed] [Google Scholar]
- 24.Odobasic D, Kitching AR, Semple TJ, Holdsworth SR. Endogenous myeloperoxidase promotes neutrophil-mediated renal injury, but attenuates T cell immunity inducing crescentic glomerulonephritis. J Am Soc Nephrol. 2007;18:760–70. doi: 10.1681/ASN.2006040375. [DOI] [PubMed] [Google Scholar]
- 25.Janssen WJ, Henson PM. Cellular regulation of the inflammatory response. Toxicol Pathol. 2012;40:166–73. doi: 10.1177/0192623311428477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Eddy AA. Interstitial macrophages as mediators of renal fibrosis. Exp Nephrol. 1995;3:76–9. [PubMed] [Google Scholar]
- 27.Hatcher H, Planalp R, Cho J, Torti FM, Torti SV. Curcumin: from ancient medicine to current clinical trials. Cell Mol Life Sci. 2008;65:1631–52. doi: 10.1007/s00018-008-7452-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nomura A, Nishikawa K, Yuzawa Y, et al. Tubulointerstitial injury induced in rats by a monoclonal antibody which inhibits function of a membrane inhibitor of complement. J Clin Invest. 1995;96:2348–56. doi: 10.1172/JCI118291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ghosh SS, Massey HD, Krieg R, et al. Curcumin ameliorates renal failure in 5/6 nephrectomized rats: role of inflammation. Am J Physiol Renal Physiol. 2009;296:F1146–57. doi: 10.1152/ajprenal.90732.2008. [DOI] [PubMed] [Google Scholar]
- 30.Kulkarni AP, Ghebremariam YT, Kotwal GJ. Curcumin inhibits the classical and the alternate pathways of complement activation. Ann N Y Acad Sci. 2005;1056:100–12. doi: 10.1196/annals.1352.007. [DOI] [PubMed] [Google Scholar]
- 31.Alexander JJ, Aneziokoro OG, Chang A, et al. Distinct and separable roles of the complement system in factor h-deficient bone marrow chimeric mice with immune complex disease. J Am Soc Nephrol. 2006;17:1354–61. doi: 10.1681/ASN.2006020138. [DOI] [PubMed] [Google Scholar]
- 32.Alexander JJ, Hack BK, Jacob A, Chang A, Haas M, Finberg RW, Quigg RJ. Abnormal immune complex processing and spontaneous glomerulonephritis in complement factor H-deficient mice with human complement receptor 1 on erythrocytes. J Immunol. 2010;185:3759–67. doi: 10.4049/jimmunol.1000683. [DOI] [PubMed] [Google Scholar]
- 33.Purkayastha S, Berliner A, Fernando SS, et al. Curcumin blocks brain tumor formation. Brain Res. 2009;1266:130–8. doi: 10.1016/j.brainres.2009.01.066. [DOI] [PubMed] [Google Scholar]
- 34.Bao L, Haas M, Kraus DM, et al. Administration of a soluble recombinant complement C3 inhibitor protects against renal disease in MRL/lpr mice. J Am Soc Nephrol. 2003;14:670–9. doi: 10.1097/01.asn.0000051597.27127.a1. [DOI] [PubMed] [Google Scholar]
- 35.Jacob A, Hack B, Bai T, Brorson JR, Quigg RJ, Alexander JJ. Inhibition of C5a receptor alleviates experimental CNS lupus. J Neuroimmunol. 2010;221:46–52. doi: 10.1016/j.jneuroim.2010.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hillegass LM, Griswold DE, Brickson B, Albrightson-Winslow C. Assessment of myeloperoxidase activity in whole rat kidney. J Pharmacol Methods. 1990;24:285–95. doi: 10.1016/0160-5402(90)90013-b. [DOI] [PubMed] [Google Scholar]
- 37.Bright JJ. Curcumin and autoimmune disease. Adv Exp Med Biol. 2007;595:425–51. doi: 10.1007/978-0-387-46401-5_19. [DOI] [PubMed] [Google Scholar]
- 38.Mito S, Watanabe K, Harima M, et al. Curcumin ameliorates cardiac inflammation in rats with autoimmune myocarditis. Biol Pharm Bull. 2011;34:974–9. doi: 10.1248/bpb.34.974. [DOI] [PubMed] [Google Scholar]
- 39.Srivastava RM, Singh S, Dubey SK, Misra K, Khar A. Immunomodulatory and therapeutic activity of curcumin. Int Immunopharmacol. 2011;11:331–41. doi: 10.1016/j.intimp.2010.08.014. [DOI] [PubMed] [Google Scholar]
- 40.Kozono Y, Kotzin BL, Holers VM. Resting B cells from New Zealand Black mice demonstrate a defect in apoptosis induction following surface IgM ligation. J Immunol. 1996;156:4498–503. [PubMed] [Google Scholar]
- 41.Mantovani A, Sica A, Locati M. New vistas on macrophage differentiation and activation. Eur J Immunol. 2007;37:14–6. doi: 10.1002/eji.200636910. [DOI] [PubMed] [Google Scholar]
- 42.Furuichi K, Wada T, Iwata Y, et al. CCR2 signaling contributes to ischemia–reperfusion injury in kidney. J Am Soc Nephrol. 2003;14:2503–15. doi: 10.1097/01.asn.0000089563.63641.a8. [DOI] [PubMed] [Google Scholar]
- 43.Kitagawa K, Wada T, Furuichi K, et al. Blockade of CCR2 ameliorates progressive fibrosis in kidney. Am J Pathol. 2004;165:237–46. doi: 10.1016/S0002-9440(10)63292-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gordon S, Mantovani A. Diversity and plasticity of mononuclear phagocytes. Eur J Immunol. 2011;41:2470–2. doi: 10.1002/eji.201141988. [DOI] [PubMed] [Google Scholar]
- 45.Gordon S. The macrophage: past, present and future. Eur J Immunol. 2007;37(Suppl. 1):S9–17. doi: 10.1002/eji.200737638. [DOI] [PubMed] [Google Scholar]
- 46.Gordon S. Alternative activation of macrophages. Nat Rev Immunol. 2003;3:23–35. doi: 10.1038/nri978. [DOI] [PubMed] [Google Scholar]
- 47.Haskill S, Becker S. Flow cytometric analysis of macrophage heterogeneity and differentiation: utilization of electronic cell volume and fluorescent substrates corresponding to common macrophage markers. J Reticuloendothel Soc. 1982;32:273–85. [PubMed] [Google Scholar]
- 48.Hill PA, Lan HY, Nikolic-Paterson DJ, Atkins RC. ICAM-1 directs migration and localization of interstitial leukocytes in experimental glomerulonephritis. Kidney Int. 1994;45:32–42. doi: 10.1038/ki.1994.4. [DOI] [PubMed] [Google Scholar]
- 49.Hill PA, Lan HY, Nikolic-Paterson DJ, Atkins RC. The ICAM-1/LFA-1 interaction in glomerular leukocytic accumulation in anti-GBM glomerulonephritis. Kidney Int. 1994;45:700–8. doi: 10.1038/ki.1994.94. [DOI] [PubMed] [Google Scholar]
- 50.Vaporciyan AA, Mulligan MS, Warren JS, et al. Up-regulation of lung vascular ICAM-1 in rats is complement dependent. J Immunol. 1995;155:1442–9. [PubMed] [Google Scholar]
- 51.Ward PA. The harmful role of c5a on innate immunity in sepsis. J Innate Immun. 2010;2:439–45. doi: 10.1159/000317194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ward PA. Role of complement, chemokines, and regulatory cytokines in acute lung injury. Ann N Y Acad Sci. 1996;796:104–12. doi: 10.1111/j.1749-6632.1996.tb32572.x. [DOI] [PubMed] [Google Scholar]
- 53.Eddy AA. Molecular insights into renal interstitial fibrosis. J Am Soc Nephrol. 1996;7:2495–508. doi: 10.1681/ASN.V7122495. [DOI] [PubMed] [Google Scholar]
- 54.Eddy AA. Molecular basis of renal fibrosis. Pediatr Nephrol. 2000;15:290–301. doi: 10.1007/s004670000461. [DOI] [PubMed] [Google Scholar]
- 55.Pawluczyk IZ, Harris KP. Macrophages promote prosclerotic responses in cultured rat mesangial cells: a mechanism for the initiation of glomerulosclerosis. J Am Soc Nephrol. 1997;8:1525–36. doi: 10.1681/ASN.V8101525. [DOI] [PubMed] [Google Scholar]
- 56.Leonarduzzi G, Scavazza A, Biasi F, et al. The lipid peroxidation end product 4-hydroxy-2,3-nonenal up-regulates transforming growth factor β1 expression in the macrophage lineage: a link between oxidative injury and fibrosclerosis. FASEB J. 1997;11:851–7. doi: 10.1096/fasebj.11.11.9285483. [DOI] [PubMed] [Google Scholar]
- 57.Okada S, Shikata K, Matsuda M, et al. Intercellular adhesion molecule-1-deficient mice are resistant against renal injury after induction of diabetes. Diabetes. 2003;52:2586–93. doi: 10.2337/diabetes.52.10.2586. [DOI] [PubMed] [Google Scholar]
- 58.Sassy-Prigent C, Heudes D, Mandet C, et al. Early glomerular macrophage recruitment in streptozotocin-induced diabetic rats. Diabetes. 2000;49:466–75. doi: 10.2337/diabetes.49.3.466. [DOI] [PubMed] [Google Scholar]
- 59.Bao L, Zhou J, Holers VM, Quigg RJ. Excessive matrix accumulation in the kidneys of MRL/lpr lupus mice is dependent on complement activation. J Am Soc Nephrol. 2003;14:2516–25. doi: 10.1097/01.asn.0000089831.96794.0b. [DOI] [PubMed] [Google Scholar]
- 60.MacKay K, Striker LJ, Stauffer JW, Doi T, Agodoa LY, Striker GE. Transforming growth factor-β. Murine glomerular receptors and responses of isolated glomerular cells. J Clin Invest. 1989;83:1160–7. doi: 10.1172/JCI113996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Suzuki S, Ebihara I, Tomino Y, Koide H. Transcriptional activation of matrix genes by transforming growth factor β1 in mesangial cells. Exp Nephrol. 1993;1:229–37. [PubMed] [Google Scholar]
- 62.McKay NG, Khong TF, Haites NE, Power DA. The effect of transforming growth factor β1 on mesangial cell fibronectin synthesis: increased incorporation into the extracellular matrix and reduced pI but no effect on alternative splicing. Exp Mol Pathol. 1993;59:211–24. doi: 10.1006/exmp.1993.1040. [DOI] [PubMed] [Google Scholar]
- 63.Yoshioka K, Takemura T, Murakami K, et al. Transforming growth factor-β protein and mRNA in glomeruli in normal and diseased human kidneys. Lab Invest. 1993;68:154–63. [PubMed] [Google Scholar]